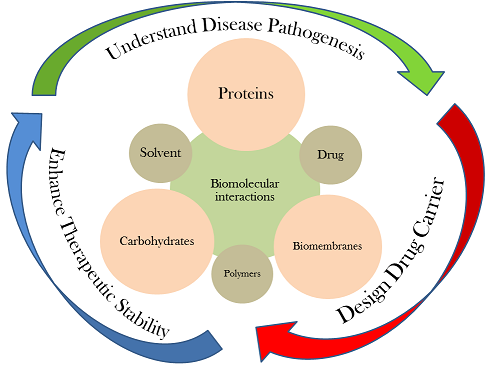
Our research revolves around interactions among biomolecules, mainly proteins, carbohydrates, biomembranes, and their interaction with small molecules such as solvent, cosolutes, and other chemical compounds (drug). We employ molecular simulation techniques like Molecular Dynamics (MD) and Monte Carlo (MC) simulations to probe the molecular level interactions and map them to the macroscopic properties through statistical mechanics principles. Many underlying processes of our interest, mainly aggregation and self-assembly, span over a wide range of length and time scales; we adopt a multi-scale modeling approach that includes atomistic simulations, coarse grained simulations, implicit solvent modeling, biased sampling techniques, and accelerated molecular dynamics for enhanced sampling of conformational space. We also employ empirical modeling, network analysis, and machine learning strategies.
We are currently focusing on the following research domains:
- Understanding the Disease Pathogenesis and Drug Design
- Design of Drug Delivery Carriers
- Assessing and Enhancing Therapeutics Stability