MultiScale Mechanics Lab
Indian Institute of Technology Madras
Research Overview

Our research interests lie at the interface of solid mechanics and materials science, with an emphasis on plasticity, environmentally assisted mechanical degradation/electrochemistry corrosion, and development of reduced-order models. The thermomechanical properties of metallic alloys are greatly influenced by the underlying microstructural features (such as grain boundaries, triple junctions, dislocations and precipitate/second phase particle distribution etc.) which spans across several length scales. Therefore, uncovering the role of these microstructural constituents can aid in the bid to design energy efficient and safer structural components. However, conventional research approaches are inadequate in accelerating the development of advanced materials. Thus, there is a need to formulate multiscale approach, where complimentary experimental and modelling efforts can aid in the deeper understanding of the role microstructural features have on the overall mechanical behavior. Therefore, we @ MML strive to develop a research program that will focus on addressing these challenges by combining experimental and modelling techniques.
Past Research Projects
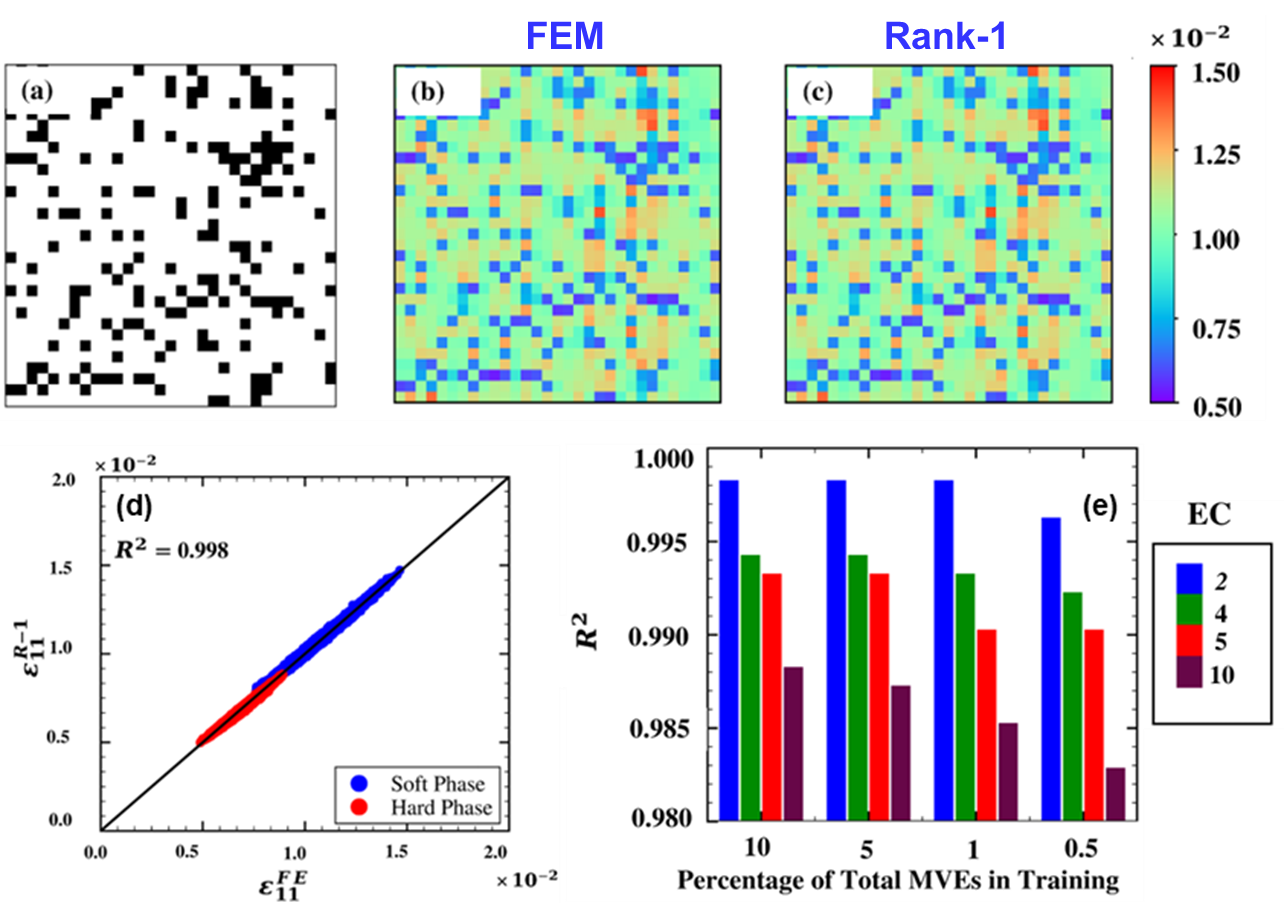
Efficient Prediction of Meso-scale Mechanical Behavior
The design of next-generation structural components demands better understanding of the mechanical variability due to the underlying heterogenous microstructure. To accurately quantify this variability a collection of a large dataset is required to account for the material heterogeneities. However, this requires tremendous computational/experimental resources. Low rank approximation (LRA) methods are part of a powerful suite of tools that enable the development of accurate reduced order models that can enable us to understand the mechanical variability introduced by the microstructure. The method describes an approximate functional form for the mechanical property of interest, which is fine tuned based on a small dataset of computational/experimental results. In a recent work, the LRA method was utilized to predict the local strain field for a heterogenous material. Interestingly, the LRA method demonstrated outstanding predictive capabilities across a range of different heterogenous materials, while being computationally efficient. In summary, the LRA method exhibited promise to accelerate solution for localization problems. These findings indicate the dawn of a new era in computational solid mechanics providing accelerated prediction based on a physics framework.
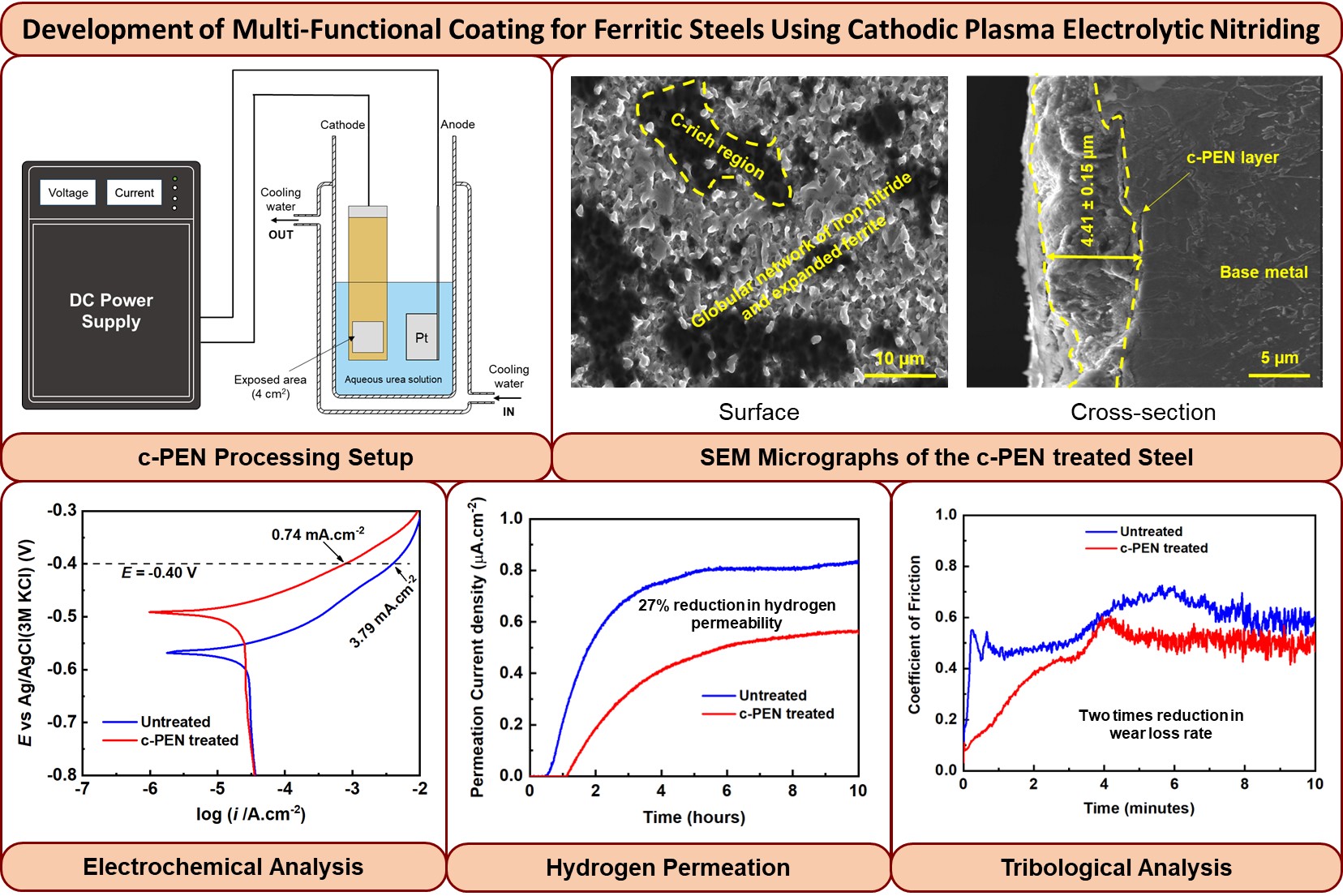
Surface Modification of Ferritic Steels for Engineering Applications
Cathodic plasma electrolytic nitriding (c-PEN) technique has been utilized to modify a low-alloy ferritic steel (2.25Cr-1Mo) surface and assess the effect of the c-PEN layer on corrosion, hydrogen permeation, and tribological behavior of the steel. The surface morphology and phase composition of the c-PEN-treated surface were analyzed, and it was found that the surface exhibits a globular network morphology of iron nitride and expanded ferrite. The potentiodynamic polarization results showed that the c-PEN treatment created an electrochemically noble surface compared to the untreated steel. Next, electrochemical hydrogen permeation experiments carried out on the nitrided surface exhibited a noticeable drop in hydrogen permeability, diffusivity, and reversible trap density of the steel. Furthermore, based on nanomechanical and tribological characterization, the c-PEN treatment was found to create a noticeably harder and wear-resistant surface. Overall, these findings demonstrate the applicability of c-PEN treatment to create a multi-functional coating for low-alloy steels that can assist in mitigating the effect of various harsh environments.
Multi-scale Assessment of the Effect of Hydrogen on Plasticity of α-Fe
Hydrogen embrittlement is an environmental phenomenon that affects both the physical and chemical properties of intrinsically ductile metals. There is a lack of comprehensive understanding on the role of plasticity during the mechanical degradation of steels when exposed to a hydrogen rich environment. Therefore, the current study employs a multi-scale approach, combining DFT, MD, and crystal plasticity techniques, to comprehensively investigate the impact of hydrogen concentration on dislocation-based plasticity in α-Fe. The findings are incorporated into a non-Schmid crystal plasticity model for accurate assessment of meso-scale plastic deformation in polycrystalline α-Fe.

Electrochemical Polarization and Mechanical Behavior of Mg based Intermetallics
The room temperature ductility and mechanical properties of Mg are usually enhanced by alloying additions. Based on the thermomechanical processing, the presence of critical concentration of alloying element typically leads to the formation of stable binary intermetallic phases with Mg thereby, distinctly altering the microscopic electrochemical and mechanical properties of the alloy. However, the secondary intermetallic phases in Mg alloys are typically of sub-micron size, thus accurate electrochemical and mechanical characterization is a challenging issue. Using first-principles calculations, the electrochemical and mechanical behavior of various Mg intermetallics was comprehensively quantified. Overall, the computational framework provides an accurate screening tool that can assist in alloy design and development of coatings.

Effect of Hydrogen on Plastic Response of Metals
Among the various non-metallic solutes which comes from environment, hydrogen plays a significant role in determining macroscopic plastic behavior of broad class of metallic systems. In order to understand the role of hydrogen on the plastic deformation of pure metallic density functional theory calculations were utilized to evaluate ideal shear strength and elastic constants of several structural metals which are key input for deriving Peierls stress of dislocation using classical Peierls-Nabaro framework. Further several experimentals and DFT calculations were utilized to develop a classical multibody interatomic potential for Fe-H system.

Effect of Mechanical Deformation on Corrosion Behavior
In general, there is a lack of fundamental understanding on the role of mechanical deformation plays in the corrosion behavior. In this work, the role of mechanical deformation on the galvanic corrosion behavior was examined across a wide range of mechanical and electrochemical conditions using a novel modeling framework. A noticeable increase in the peak pit depth was observed due to the onset of plastic deformation. Furthermore, the presence of tensile loads was found to increase the tendency for localized corrosion. Overall, the findings presented here highlight the complex interactions that occur between the mechanical and electrochemical processes during stress assisted corrosion of galvanic joints.

Hydrogen Embrittlement
Hydrogen embrittlement (HE) is a phenomenon that affects both the physical and chemical properties of several intrinsically ductile metals. In this work, the effects of hydrogen on the defect interaction during mechanical deformation was examined using a multiscale perspective. For instance, the grain boundaries (GBs) act as preferential site for hydrogen aggregation eventually leading to intergranular failure. Therefore, we thoroughly examined a large database GBs to identify GBs that were immune to hydrogen segregation. GBs present an effective barrier to dislocation motion, thereby strengthening the material. The understanding of the interactions between the GBs and dislocations in a hydrogen rich environment is critical to gain insights into the events that lead to intergranular crack initiation. Therefore, we employed molecular dynamics to study the interactions between screw dislocations and several <111> tilt GBs in Fe. The segregation of adequate hydrogen along the GB was found to increase the resistance offered by the GBs to dislocation motion, thereby increasing the dislocation pile up size that can finally lead to an accumulation of sufficient strain energy to cause intergranular crack initiation.

The Role of Triple Junctions on the Structural Stability of NC materials
Nanocrystalline (NC) metals (mean grain sizes (d) ≤100 nm) have enhanced mechanical strength as compared to coarse-grained metals (d ≥ 1 mm), thus are a promising alternative as structural materials for future high energy nuclear reactors. However, during extreme conditions, the NC microstructure has been found to be thermodynamically unstable, thereby limiting its applicability. For small grain sizes (< 10 nm), the triple junctions (TJs) have been observed to have a significant contribution on the material behavior. Using atomistic simulations, we demonstrated that the strain energy evolution around the TJ can provide insights into the distinct segregation behavior of point defects and solute atoms. Next, the activation energy for vacancy diffusion was found to linearly increase for thermodynamically stable TJs, i.e. the TJs that had lower resolved line tension. Interestingly, the examination of solute binding behavior revealed a localized region of stable sites around the TJs aids in accommodation of high solute concentration at high temperatures.

Improving the Formability of Mg Alloys
In wrought magnesium alloys, the room-temperature plasticity is largely controlled by the limited number of active slip systems such as basal slip and extension twinning. The insufficient number of active slip systems therefore limits the broader structural applicability of Mg-alloys. Using first-principles calculations, we investigate the effects of several different alloying elements on the ideal shear resistance across various slip systems of Mg. The results reveal that the addition of a Ce, Y or Zr solute atom, decrease the ideal shear resistance; whereas, a substitution of a Sn, Li, Al or Zn atom, respectively, increases the ideal shear resistance of Mg. The electronic density of states and valence charge transfer calculations can explain the profound effect of various solutes on the shear resistance of Mg. Finally, this understanding could enable the development of Mg alloys with improved room temperature formability for structural applications.

The Role of Nitrogen on Hydride Formation in pure Nb
The absorption of hydrogen and subsequent hydride precipitation degrades the superconducting properties of Nb. The addition of various dopant elements, particularly nitrogen have shown to cause a decrease in hydride concentration. Nonetheless, the underlying mechanisms associated with kinetics of hydrogen and the thermodynamic stability of hydride precipitates are not well known. the presence of nitrogen significantly increased the energy barrier for hydrogen diffusion from one tetrahedral site to another interstitial site.
